The general engineering simulation software WELSIM has released the latest 2025R1 version (internal version 3.0). Compared to the previous version, the 2025R1 version includes many new features and enhancements, providing better support for various types of engineering simulation CAE analyses, especially with the addition of molecular dynamics analysis capabilities.

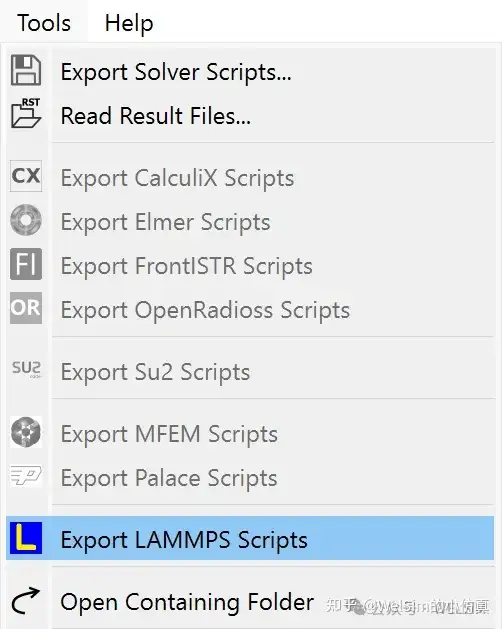
Support for the Molecular Dynamics Solver LAMMPS
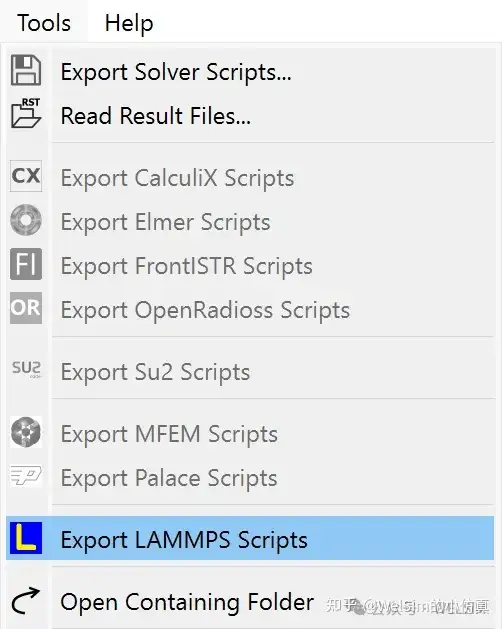
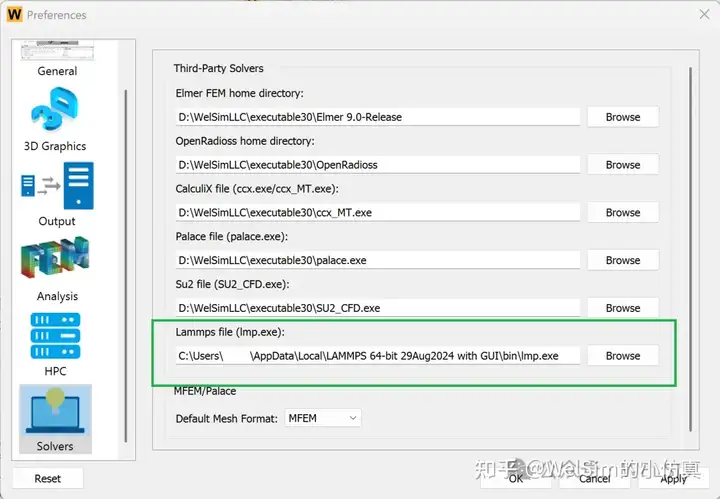
The new version adds pre- and post-processing modules for the LAMMPS solver. Users can quickly generate the input scripts required for LAMMPS computation and can also directly call LAMMPS program through WELSIM for solving.
Currently, 36 commands are supported, covering approximately 36% of all LAMMPS commands, and all of these are core commands, which can meet the needs of general analyses.
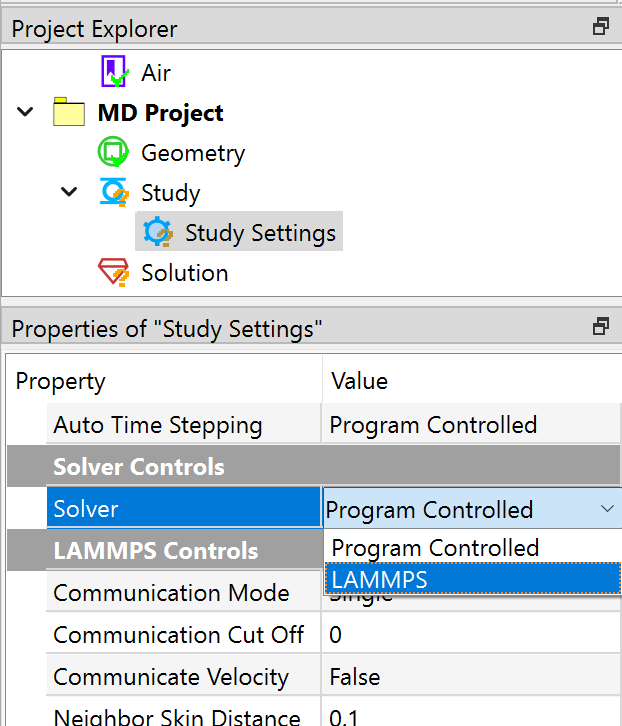
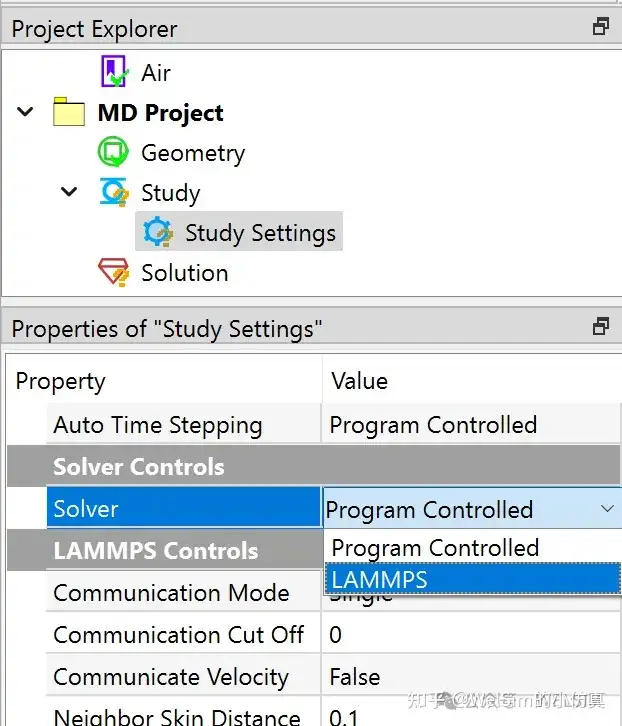
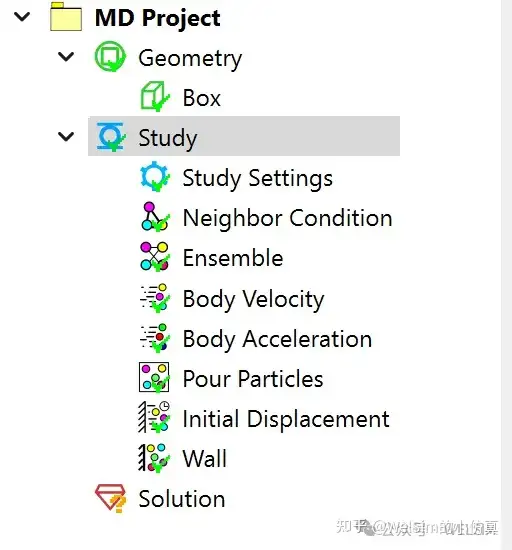
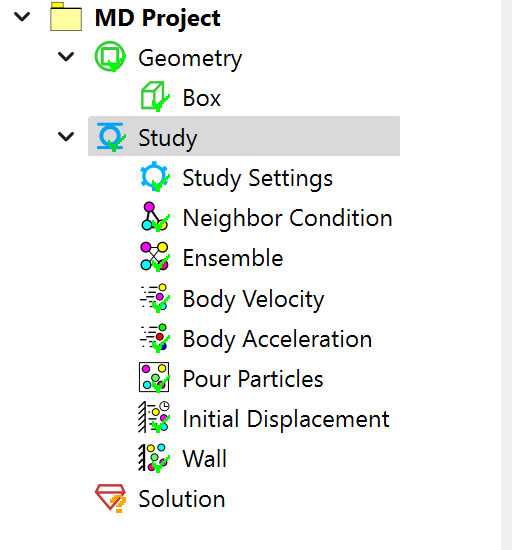
In the pre-processing module, a molecular dynamics project option has been added, along with various conditions such as particle neighbor conditions, particle velocity, particle acceleration, initial displacement, boundary walls, force fields, etc. These pre-processing objects and properties are intuitive and easy to use.
The solving feature is consistent with the finite element solving method. During the computation, the solver’s output information is displayed in a pop-up window, allowing users to monitor the calculation status.
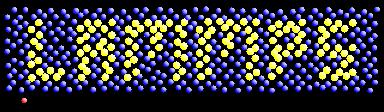
In terms of post-processing, WELSIM also supports some molecular dynamics-related features. It can read the log file (log.lammps) and result files (dump.lmps) generated by LAMMPS solver and display the results. The newly added particle display module can efficiently render particle trajectories and colors.

Enhancements and Upgrades
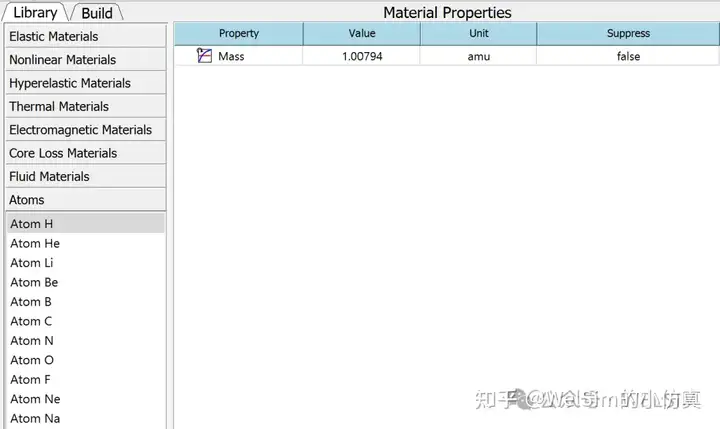
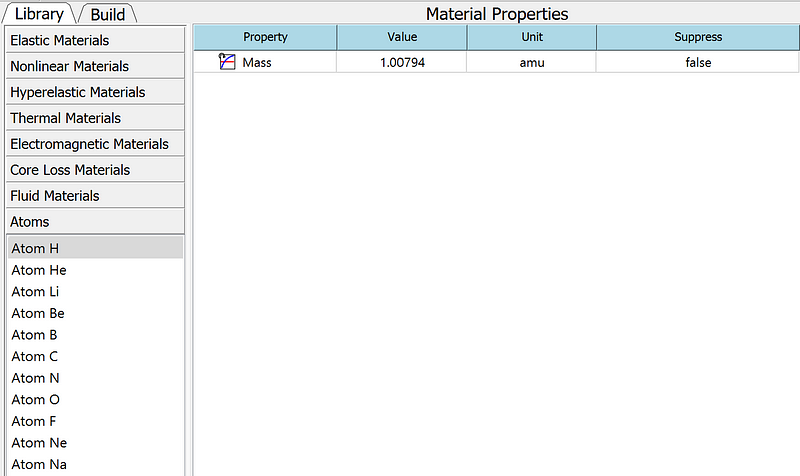
Additionally, the new version includes other new features and improvements. It supports the 3DConnexion 3D space mouse hardware and the export of result files in TecPlot format. It also adds atomic mass units and basic atomic materials such as H, He, Li, Be, B, C, N, O, F, Ne, and Na.
WELSIM and the author are not affiliated with LAMMPS, TecPlot, or 3DConnexion. The use of LAMMPS, TecPlot, and 3DConnexion is for reference in this technical blog post and software usage only.